Most Frequently Asked Pharma Interview Questions
1.What is the definition of SOP?
SOPs are detailed written instructions for the operations routinely performed in the course of any activities associated with pharmaceutical manufacturing.
Or
A written authorized procedure which gives instructions for performing operations not necessarily specific to a given product / material, but of a more general nature the equipments preventive maintenance and cleaning; recall of products; purchasing; cleaning of premises and environmental control; sampling and inspection etc.
Or
These are guidelines which describe how the activity is to be performed. To achieve uniformity of results by each individual, it is mandatory to follow these guidelines. SOP is like a “TELL and SHOW” concept.
Tell – means to establish and teach how the activity is to be carried out.
Show – means to provide the documented proof for the activity carried out.
2.What are the contents of the SOP?
Objective/Purpose, Scope, Responsibility, Accountability, Procedure, List of formats/Annexure, Abbreviations, Reference, Revision History
3.Which information should master document carry on every page not just one of the pages to meet GMP?
Page number, document reference number and authorizing signatures
4.How many SOPs required for equipment and what are those?
Operation, Cleaning, Preventive maintenance/ Calibration, Sampling procedure
5. What is the Batch production and control record (BPCR)?
BPCR are prepared for each intermediate and API and include the complete information relating to the completion of each significant step in the Batch production.
6.What is the Master production & control record (MPCR)?
To ensure the uniformity from batch to batch, master production instructions for each intermediate and API are prepared, dated and signed by one person, immediately checked, dated and signed by a person in the quality unit.
7.What are the content of the MPCR?
∙ The name of the intermediate or API being manufactured and an identifying document reference code, if applicable
∙ A complete list of raw materials and intermediates designated by names or codes sufficiently specific to identify any special quality characteristics.
∙ An accurate statement of the quantity or ratio of each raw material or intermediate to be used, including the unit of measure. Where the quantity is not fixed, the calculation for each batch size or rate of production should be included. Variations to quantities should be included where they are justified
∙ The production location and major production equipment to be used
∙ Detailed production instructions, including the:
– Sequences to be followed
– ranges of process parameters to be used
– sampling instructions and in-process controls with their acceptance criteria, where appropriate
– time limits for completion of individual processing steps and/or the total process, where appropriate
– expected yield ranges at appropriate phases of processing or time
-Where appropriate, special notations and precautions to be followed, or cross references to these
−The instructions for storage of the intermediate or API to ensure its suitability for use, including the labeling and packaging materials and special storage conditions with time limits, where appropriate.
8.What is the list SOPs required in QA department?
SOP for SOP, SOP for format preparation, change control, deviation, Non-conformance products, market complaints, product recall, returned goods, vendor qualification, preparation of BPCR & MPCR, Assigning of Mfg. date & Expiry date, annual product review, corrective action & preventive action, process validation, cleaning validation, equipment qualification, glossary of terms, document control, Review of BPCR & analytical test report, batch numbering system, labeling practice, personnel training, BPCR issue and retrieval, batch release, self inspection (internal audit), file numbering system, preparation of organo-gram, preparation of COA, specimen signatures, Reprocess & rework of intermediates / API, Job responsibilities, Technology transfer, measurable quality objectives etc
9.What is the difference between intermediate and drug substance (API)?
Intermediate: A material produced during steps of the processing of an API that undergoes further molecular change or purifications before it become an API (Reference: ICH Q7A). API: Any substance or mixture of substances intended to be used in the manufacturing of a drug (medicinal) product and that when used in the production of a drug, becomes an API of the drug product. Such substances are intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment or prevention of disease or to affect the structure & function of the body (Reference: ICH Q7A).
10.What is the difference between drug substance and drug product?
Drug substance (API): Any substance or mixture of substances intended to be used in the manufacture of a drug (medicinal) product and that, when used in the production of a drug, becomes an active ingredient of the drug product. Such substances are intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment, or prevention of disease or to affect the structure and function of the body (Reference: ICH Q7A).
Drug product: The dosage form in the final immediate packaging intended for marketing (Reference: ICH Q7A).
11.What is the clean room?
Clean rooms are defined as especially constructed, environmentally controlled enclosed spaces with respect to airborne particulates, temperature, humidity, air pressure, air flow patterns, air motion, vibration, noise, viable (living organisms) and lighting.
Particulate control includes:
∙ Particulate & microbial contamination
∙ Particulate concentration & dispersion
12.What are the classifications of clean rooms?
Generally clean rooms are classified in to the following types as per different guidelines: Schedule M: Grade A, Grade B, Grade C, Grade D
USFDA (US 209E): Class 1, Class 10, Class 100, Class 1000, Class 10000, Class 100,000 WHO 2002: Grade A, Grade B, Grade C, Grade D
EU GMP: Grade A, Grade B, Grade C, Grade D
ISO 14644-1: ISO-3, ISO-4, ISO-5, ISO-6, ISO-7, ISO-8, ISO-9
Britian (BS 5295): Class C, Class D, Class E or F, Class G or H, Class J, Class K
Australia (AS 1386): 0.035, 0.35, 3.5, 35, 350, 3500
Germany (VDI 2083): 1, 2, 3, 4, 5, 6
13.What is the difference between GMP & cGMP?
GMP: GMP is the part of Quality assurance which ensures that products are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the marketing authorization.
GMP are aimed primarily at diminishing the risks inherent in any pharmaceutical production. Such risks are essentially of two types:
- Cross-contamination (in particular of unexpected contamination)
- Mix-ups (confusion)
cGMP: Current Good Manufacturing Practices. This means any procedure / system adopted by the manufacturer which proves to be necessary and important for identity, strength and purity of a product.
14.What is the difference between Qualification and Validation?
Qualification is equipment / instrument oriented but validation is process oriented.
15. What is the definition of Validation?
Validation is the documented program that provides a high degree of assurance that a specific process, method or system will consistently produce a result meeting predetermined acceptance criteria.
16.What is the definition of Qualification?
Qualification is the action of proving and documenting that any equipment or ancillary systems are properly installed, work correctly, actually leads the expected results. Qualification is part of validation, but the individual qualification steps alone do not constitute process validation.
17. What are the types of validation?
Process validation, Analytical method validation, cleaning validation, facility validation, Utility validation & software validation
18.Definition of process validation and types of process validation?
Process validation is the documented evidence that the process, operated within established parameters, can perform effectively and reproducibly to produce an intermediate / API meeting
its pre-determined specifications and quality attributes.
Process validation is three types:
- Prospective process validation
- Concurrent process validation
- Retrospective process validation
- What is the prospective, concurrent and retrospective validation?
Prospective process validation: Prospective Process validation shall be carried out for all the intermediate stages and Active Pharmaceutical Ingredients prior to the distribution of a new product. [ICH: GMP, EU: GMP, PIC/S: GMP]
Concurrent process validation: Any validated process undergoes a change either for the equipment or addition, deletion of a critical manufacturing process step, scale up or scale down, the same needs to be validated concurrently.
The validation is carried out only after a change of an existing validated process to support the change made or involve with the requirements.
Or
A subset of prospective validation in which API batches are released for distribution, based on extensive testing, before completion of process validation. Once data from additional batches produced under replicated conditions show uniformity, the process may be considered validated
Or
Concurrent validation can be conducted when data from replicate production runs are unavailable because only a limited number of API batches have been produced, API batches are produced infrequently, or API batches are produced by a validated process that has been modified. [ICH: GMP, EU: GMP, PIC/S: GMP]
Retrospective process validation: Validation of a process for a product already in distribution based upon accumulated production, testing and control data. [ICH: GMP, EU: GMP, PIC/S: GMP]
20.What do you mean by validation protocol and its contents of process validation?
A written plan stating, how validation will be conducted and defining acceptance criteria e.g: The protocol for manufacturing process identifies process equipments, critical process parameters, and / or operating range, product characteristics, sampling, test data to be collected, number of validations runs and acceptance test results.
Contents:
- ∙ Protocol Approval
- ∙ Table of contents
- ∙ Objective
- ∙ Scope
- ∙ Responsibility
- ∙ Accountability
- ∙ Validation team
- ∙ Brief manufacturing process (Description, Flow chart, Reaction scheme)
- ∙ Selection of batches
- ∙ List of equipments used in the manufacturing process
- ∙ List of raw materials used in the manufacturing process
- ∙ Critical operations with justification
- ∙ In-process controls with acceptance criteria
- ∙ Sampling & testing plan with frequency
- ∙ Stability programm
- ∙ Data to be complied
- ∙ Acceptance criteria
- ∙ Intermediate & final products quality & yield
- ∙ Stability specification
- ∙ Document review
- ∙ Conclusion
- ∙ Revalidation criteria
21.What is the definition of the procedure?
A documented description of the operation to be performed, the precautions to be taken, and measures to be applied directly or indirectly related to the manufacture of an intermediate / API (Reference: ICH Q7A).
22.What is the master document?
Master document is a formally authorized source document relating to specifications, and / or manufacturing / analytical methods, which is protected from un-authorized access or amendment.
∙ Documents required describing the quality system requirements in the organization. ∙ Documents required describing the process or product characteristics.
∙ Documents required by various regulatory agencies as part of compliance to GMP requirements.
∙ Documents required for legal/ regulatory supports of the organization to meet the local regulations.
∙ Any other documents required by government / regulatory agency.
23.What is documentation?
All the written production procedures, instructions and records, quality control procedures and recorded test results involved in the manufacturing of a medicinal product.
24.What is the Technology Transfer?
In the pharmaceutical industry, “technology transfer” refers to the processes that are needed for successful progress from drug discovery to product development to clinical trials to full scale commercialization or it is the process by which a developer of technology makes its technology available to commercial partner that will exploit the technology.
To assure the drug quality, it is desire to make sure 5 W’s and 1 H, that is what1, when2, and why3information should be transferred to where4and by whom5and how to transfer, then share knowledge and information of the technology transfer each other between stake holders related to drug manufacturing.
25.What are the names of different countries of GMP guidelines for manufacturing of API?
- WHO GMP – Geneva
- ICH Q7A – Europe, Japan & US
- EU GMP – Europe
- MCC – South Africa
- APIC GMP – Active Pharmaceutical Ingredient Committee (A sector group of CEFIC)
- USFDA GMP – United States of America
- PIC/S GMP- Germany
- Schedule M – Indian
26.What is preventive maintenance?
It is periodic inspection and minor repairs of equipment as per schedule given in the SOP. This enables smooth operation and long life of the equipment. It also avoids major breakdown of the equipment during manufacturing of the product.
There are two types of maintenance.
Preventive maintenance: Schedule maintenance before any break down of machinery which prevents the machine break down.
Breakdown maintenance: Maintenance was done after stopping machine breakdown. Weekly, Monthly, Quarterly, Half yearly and Yearly preventive maintenance
27.What do you mean by “Quality Assurance”?
The sum total of the organized arrangements made with the objects of ensuring that all APIs are of the quality required for their intended use and the quality systems are maintained.
28. What are the types of different training programs?
- Induction training
- Job oriented training
- cGMP training
- On-going training
29.What is cGMP?
Current Good Manufacturing Practices. This means any procedure / system adopted by the manufacturer which proves to be necessary and important for identity, strength and purity of a product.
30.What are the requirements for the equipment used in the manufacturing of process of API?
Material of construction used for equipment should not
∙ React with component
∙ Get corroded, cause rusting
∙ Impart any impurities, absord
∙ Should be of appropriate design, adequate size and have smooth surface.
31. How are cGMP implemented?
Training, compliance to SOPs, control on operations, following procedures / systems, monitoring through compliance audits.
32.What is solvent?
An organic or inorganic liquid used as a vehicle for the preparation of solutions or suspensions in the manufacturing of an intermediate / API.
33.What are the classifications of residual solvents?
Residual solvents are classified into three class based on the possible risk to human health: Class-I (Solvents to be avoided)
Class-II (Solvents to be limited)
Class-III (Solvents with low toxic potential)
34.What is the difference between Responsibility and Accountability?
Responsibility: Personnel directly associated with the implementation of the procedure Accountability: Person directly associated with the implementation of the system under which the procedure falls.
35.Write the names of the different countries regulatory body (Like for India, USA, UK, Australia, South Africa, Brazil, Hungary, Germany, Philippines etc.) ?
India – Schedule M
United Status of America – USFDA (United state Food and Drug Administration) Australia – TGA (Therapeutic Goods Administration)
United Kingdom – MHRA (Medicines & Health care products Regulatory Agency) South Africa – MCC (Medicine Control Council)
Brazil – ANVISA (Brazilian Health Surveillance Agency or National Sanitary Surveillance Agency) Hungary – PIC/S (Pharmaceutical Inspection Convention or Pharmaceutical Inspection Co operation Scheme)
Germany – NIP (National Institute of Pharmacy)
Philippines – BFAD (Beaureu of Food & Drug)
36.What is the abbreviation of MSDS and how many contents are mentioned & what are those?
MSDS means Material Safety Data Sheet and it contains 16 contents. Those are given below
1. Product IdentificationComposition / Information on Ingredients
- Hazards identification
- First Aid measures
- Fire fighting measures
- Accidental release measures
- Handling & storage
- Exposure controls / Personal protection
- Physical & Chemical properties
- Stability & Reactivity
- Toxicological information
- Ecological information
- Disposal consideration
- Transport information
- Regulatory information
- Other information
37.What is the static electricity?
Denoting / pertaining to electricity which is at rest. The electricity which is present on surface of a non-conductive body, where it is trapped from escaping, is called static electricity.
38. What is the different types of Qualifications and write its flow?
Qualifications are as follows: Design Qualification, Installation Qualification, Operational Qualification, and Performance Qualification.
URS/DS —–FAT—–SAT—–DQ—–IQ—–OQ—–PQ
39.What is audit/inspection and Why quality audit? Write different types of audits/inspection?
A planned and systematic examination and check of a system, procedure or operation in order to monitor compliance with and the effectiveness of established standards and to allow for improvement and corrective measures where required.
Quality audit because of:
∙ To assess the effectiveness of the quality management system
∙ Assessing conformance
∙ Investigating problems
∙ Continual improvement of performance
∙ Assessing for Registration
∙ Reducing cost of operation
∙ Legal requirement
Types: 1. Study/test based inspection
- Facility based inspection
- Process based inspection
40.Why nitrogen gas used in the manufacturing area at room temperature and why not other gas?
Because of nitrogen is chemically less reactive and does not react with other elements at ordinary temperature. It is due to strong bonding in its molecule.
41.What are the different types of cleanings?
There are three types of cleanings:
∙ Batch to Batch cleaning
∙ Periodically cleaning
∙ Product change over cleaning
42.What is blending?
Blending is defined as the process of combining materials within the same specification to produce a homogeneous intermediate or API.
43.What is expiry date & re-test date?
Expiry date: The date place on the container / labels of an API designated the time during which the API is expected to remain within established shelf life specifications if stored under defined conditions and after which it should not be used.
Re-test date: The date when a material should be re-examined to ensure that it is still suitable for use. The period of time during which the drug substance is expected to remain within its specifications and therefore, can be used in the manufacturing of the drug product, provided that drug substance has been stored under the defined conditions.
44.What is difference between reprocess & rework?
Reprocess: Introducing an intermediate or API, including one that does not conform to standards or specifications, back into the process and repeating a crystallization step or other appropriate chemical or physical manipulation steps (e.g., distillation, filtration, chromatography, and milling) that are part of the established manufacturing process. Continuation of a process step after an in-process control test has shown that the step is incomplete, is considered to be part of the normal process, and is not reprocessing.
Reworking: Subjecting an intermediate or API that does not conform to standards or specifications to one or more processing steps that are different from the established manufacturing process to obtain acceptable quality intermediate or API (e.g., recrystallizing with a different solvent).
45.What is deviation & its types?
Deviation is departure from the approved instructions /established standards. There are two types of deviation and given below:
Controlled / planned deviation: Any deviation from documented procedure opted deliberately for temporary period to manage unavoidable situation or improving the performance of the operations, without affecting the quality & yield of drug substance and safety of the operations shall be termed as controlled / planned deviation.
Uncontrolled / unplanned deviation: Any deviation occurred in unplanned or uncontrolled manner such as system failure or equipment breakdown or manual error shall be termed as uncontrolled / unplanned deviation.
46.What is change control and its types?
Change control is a system that control change by
- Identifying ownership of the change
- Allowing for review and approval of the change.
iii. Preventing changes that could adversely affect product quality or conflict with registration or regulatory requirement.
- Providing an assessment of change and monitors the impact of change.
Level 1 (Minor): Are those that are unlikely to have any detectable impact on the quality attributes of the product.
Level 2 (Major): Are those that are likely to have a significant impact on the quality attributes of the product.
The type of reasons for change control:
– Regulatory requirement
– GMP implementation / enhancement
– Quality improvement
– Capacity enhancement
– Introduction of new product in existing facility
– Cost reduction
– Automation
– Aging of facility
– To manage the unavoidable situation
– Market requirement
47.What is contamination and cross-contamination?
Contamination: The undesired introduction of impurities of a chemical or Microbiological nature, or of foreign matter, in to or onto a raw material, intermediate, or API during production, sampling, packaging or repackaging, storage or transport.
Cross-contamination: Contamination of a material or of a product with another material or product.
48.What is Batch number and batch?
Batch Number: A unique combination of numbers, letters, and/or symbols which identifies a batch (or lot) and from which the production and distribution history can be determined Batch: A specific quantity of material produced in a process or series of processes so that it is expected to be homogeneous within specified limits. In the case of continuous production, a batch may correspond to a defined fraction of the production. Batch size may be defined either by a fixed quantity or the amount produced in a fixed time interval.
49.What is quarantine?
The status of materials isolated physically or by other effective means pending a decision on their subsequent approval or rejection.
50.What is definition of critical process parameters?
A process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired quality.
51. What is mother liquor?
The residual liquid which remains after the crystallization or isolation processes. Mother liquor may contain un-reacted materials, intermediates, levels of the API and/or impurities. It may be used for further processing.
52.What is the difference between theoretical and expected yield?
Theoretical yield: The quantity that would be produced at any appropriate phase of production, based upon the quantity of material to be used, in the absence of any loss or error in actual production.
Expected yield: The quantity of material or the percentage of theoretical yield anticipated at any appropriate phase of production based on previous laboratory, pilot scale, or manufacturing data
53.What is OOS?
Out of Specification (OOS) results are those results, generated during testing that do not comply with the relevant specification or standards or with the defined acceptance criteria.
54. What is CAPA?
CAPA is the Corrective Action & Preventive Action.
Corrective Action: Action taken to eliminate the causes of an existing non-conformity, defect or other undesirable situation to prevent recurrence. [Actions taken after the occurrence of a defect or problem to stop the same from recurrence].
Preventive Action: Action taken to eliminate the causes of potential non-conformity, defect or other undesirable situation to prevent occurrence. [Actions initiated before the occurrence of a defect or problem to prevent the same occurrence].
55.What is the ICH?
Write its aim/purpose and names of the different parties & different regions? ICH means “International conference on harmonization”.
Aim/Purpose: “Ensure good quality, safety and effective medicines are developed and registered in the most effective manner, through harmonization of technical requirements” Different Parties:
- European commission – European Union (EMEA)
- European Federation of Pharmaceutical Industries & Association (EFPIA)
- Minister of health, Labour & Welfare, Japan (MHLW)
- Japan Pharmaceutical Manufactures Association (JPMA)
- US Food & drugs Administration (FDA)
- Pharmaceutical Research & Manufactures of America (PhRMA)
Different regions:
- European Union (EMEA)
- United states of America (USFDA)
- Japan (MHLW)
56.What URS, DS, FAT, SAT, DQ, IQ, OQ, PQ?
URS:
DS:
FAT:
SAT:
DQ:
Installation Qualification (IQ): Establishing a high degree of confidence that the equipment as installed is consistent with manufacture’s requirements and specifications.
Operating Qualification (OQ): Establishing a high degree of confidence that the equipment as installed is able to consistently operate within established limits and tolerances. Performance Qualification (PQ): Establishing a high degree of confidence, with appropriate testing that the equipment, under normal operating conditions, will consistently produce a quality product.
57.Difference between validation & testing?
Both are not same. Testing is defined as the identification of errors (difference between expected & actual results) in a system. Validation is defined as documented evidence that a system performance as expected. Validation includes testing but it is more – for instance, checking the documents for completeness & correctness.
58.Why water is used extensively as a coolant in heat exchange equipments?
Because of the abundance and high heat capacity, water is used as coolant in heat exchange equipment.
59.What are the different characteristics of the fluid are to be considered while deciding its route in a heat exchanges?
The following characteristic of the fluid are to be considered while deciding its route in a heat exchanger: a) Viscosity b) Fouling c) Corrosiveness d) Pressure
60.When steam distillation recommended?
a) To separate appreciable quantities of higher boiling materials.
b) To separate relatively small amounts of volatile impurity from a large amount of material.
c) Where use of direct-fired heaters is detrimental to the materials.
d) Where the material is to be subjected to distillation is thermally unstable or will react with other component associated with it at the boiling temperature.
e) Where the material cannot be distilled by in-direct heating even under low pressure because of the high boiling temperature.
61.What is the difference between instrument & equipment?
Instrument: A device that takes a physical measurement and displays a value or has no control or analytical functions. e.g.: Stop watch, timers & thermometer.
[A device <chemical, electrical, hydraulic, magnetic, mechanical, optical, pneumatic> used to test, observe, measure, monitor, alter, generate, record, calibrate, manage or control physical properties, movements, or other characteristics].
Equipment: A device or collection of components that perform a process to produce a result. [The collective analytical measurement instruments in conjunction with firmware, assembled to perform a mechanical process]
62.What is HVAC?
The HVAC is designed to circulate the air in the area after passing it over cooling & heating coils to maintain the required environmental conditions & passing it through the series of filters to maintain desired cleanliness level in the area. The air in-take and out-take of the system is designed to maintain certain degree of pressure gradient in the area as per requirements.
Or
HVAC system function is to condition (heating & cooling), replace (makeup, fresh air, oxygen replacement), and pressurize (contaminant) and clean (filter) the air in the environment to meet the required operational conditions.To achieve this objective, electrical, mechanical & electronic components are arranged in several configurations such that they produce the expected results.
63.What is the meaning of Q, S, E, M in the ICH?
“Q” stands for Quality; “S” stands for Safety, “E” stands for Efficacy and “M” stands for Multi dispensary
64.How many guidelines are present in Q & what are those, describe in detail?
In Quality (Q), total 10 guidelines are present. Those are as follows:
- Q1 – Stability
- Q2 – Analytical Method validation
- Q3 – Impurities
- Q4 – Pharmacopoeia
- Q5 – Biotechnological quality
- Q6 – Specification
- Q7 – Good Manufacturing Practice (GMP)
- Q8 – Pharmaceutical Development
- Q9 – Quality Risk Management
- Q10 – Pharmaceutical Quality System
65.How many types of raw material and packing material?
Raw materials are classified into two types. Those are as follows:
- Key raw material
- Other raw material
Packing materials are classified into two types. Those are as follows:
- Primary Packing material
- Secondary Packing material
66.Define the Key raw material/ starting material & primary packing material?
Key raw material/starting material:
Starting material shall be defined as that which is
∙ Incorporated as a significant structural fragment of the API / Drug Intermediate and ∙ Having significant effect on the Quality and Yield of the product.
∙ Starting material shall be identified in TDP.
Primary Packing material: Packing material, which come in direct contact with the API/Intermediate are considered as Primary packing material.
67.What is cleaning validation?
Cleaning validation is documented evidence that an approved cleaning procedure will provide equipment which is suitable for processing of pharmaceutical products or APIs. Cleaning validation is the confirmation of reliable cleaning products so that the analytical monitoring may be omitted or reduced to a minimum in the routine phase.
It describes the validation of cleaning procedures for the removal of contaminants associated with the previous products, residues of cleaning agents as well as the control of potential microbial contaminants.
68.What are the sampling techniques used in the cleaning validation?
Swab sampling: Areas which are reasonably accessible & hardest to clean can be evaluated, leading to level of contamination or residue per gives surface area.
Rinse sampling: Large areas or parts of equipments which could not be swabbed should be rinse sampled or directly extracted by solvent. Tubes, nozzles, pipes or containers with surface those are not reasonably accessible for direct surface sampling have to be rinsed with solvent. In addition, inaccessible areas of equipment that cannot be routinely disassembled can be evaluated.
69.What parameters considered during performance qualification of HVAC?
The following parameters are to be considered during the performance qualification of HVAC:
- Calibration test certificates of instruments
- Training records of validation team
- Pressure drop across the HEPA & fine filters
- Air velocity measurement & calculation of Air changes
- Integrity test of HEPA filter
- Differential pressure test
- Temperature & Relative Humidity test
- Air flow direction test
- Cleanliness class verification (Non-viable particle count)
- Sound level test
- Light level test
- Air borne viable particle monitoring
- Recovery Study
70.What are the contents in the BPCR?
BPCR contains the following contents, but not limited:
- Product Name
- Stage
- BPCR Document Number
- MPCR Reference Number
- Batch Numbe
- Date of Manufacturing
- Date of Expiry/Re-test
- Batch release details
- List of equipments used
- List of raw materials & Quantity with UOM
- General instructions, Control & Safety instructions
- Detailed step wise written manufacturing procedures
- Actual results record for critical process parameters
- Identity of In-process & Laboratory test results
- Signatures of person performing details along with supervising details
- Description of Packaging details
- Yield calculation
- Representative of labels for intermediates / raw materials
- Deviation details
- Batch starting & completion date
71.What is OOT and define?
“OOT” stands for Out Of Trend. It means any test results obtained for a particular batch that is markedly different the results of the batches in a series obtained using a same validated method.
72.How will you prevent cross-contamination between two different products manufactured in the one production block?
By maintaining the proper pressure differential between the rooms with two Air handling units (if re-circulation) / one Air handling unit (if 100% fresh air)
73.What is limit of Temperature and relative humidity in the pharma area?
Temperature: 25±2˚C & Relative Humidity: 50±5%
74.What is the difference between dedicated and non-dedicated equipments?
Dedicated equipment: It is used solely for the production of a single product or product line. Concerns over cross-contamination with other products are markedly reduced. Dedicated equipments must be clearly identified with the restrictions of use in order to prevent potential errors during cleaning and preparation.
Non-dedicated equipment: Where the same piece of equipment is utilized for a range of products formulations. The prevent of cross-contamination between products becomes the main objective in the cleaning validation effort. Clearly, cleaning non-dedicated equipments represents a more significant obstacle to overcome.
75.Which instrument is used for the measuring of RPM?
Techo meter is used for the measurement of RPM.
76.Why three batches consider for the validation?
Because of First one is for information, Second one is for confirmation and Third one is for evidence.
77.If one batch is failed during the validation, then what will you do for completion of validation?
When a quality parameter fails with respect to the specification, a deviation report shall be raised and the investigation shall be conducted immediately for the identification of failure.
∙ If the reason for failure is identified, one more consecutive batch shall be considered for the validation run by taking preventive actions to avoid those failures (If necessary revise the MPCR and BPCR).
∙ If the reason is unidentified, another three consecutive batches shall be taken for validation
78.What are specifications of Purified water as per any pharmacopoeia?
Tests | Ph. Eur. |
Description | Clear, colorless liquid |
Acidity /Alkalinity | The solution is not colored red/The solution is not colored blue. |
Oxidisable substances | The solution remains faintly pink |
Chlorides | The solution shows no change in appearance for at least 15 min |
Sulphates | The solution shows no change in appearance for at least 1 hour |
Ammonium | Maximum 0.2 ppm. |
Calcium and magnesium | A pure blue colour is produced. |
Residue on evaporation | Maximum 0.001 per cent |
Aluminum | Maximum 10 ppb, |
Nitrates | NMT 0.2 ppm |
Heavy Metals | NMT 0.1 ppm |
Conductivity (At 25˚C) | NMT 5.1ms.cm-1 |
Total viable aerobic count | NMT100 CFU /ml |
Pathogens : E. coli Salmonella Pseudomonas Staphalococcus aureus | Absent Absent Absent Absent |
79.Write the different storage conditions as per any guidelines (specify the name of guideline)?
The different storage conditions are given below as per USP:
Freezer : -25°C to -10°C
Cold : Any temperature not exceeding 8°C
Refrigerator : Between 2°C and 8°C
Cool : 8°C to 15°C
Room temp. : The temperature at prevailing working area.
CRT : 20°C to 25°C
Warm : 30°C to 40°C
Excessive heat : Above 40°C
80.What is Fumigation?
81.Write the names of some fumigation agents?
82.Write the different types of fires, which are generally used in the pharmaceutical industry?
a) Chemical fire
b) Electrical fire
c) Metal fire
d) General fire
e) Gaseous fire
83.What is ISO 9001, ISO 14001, ISO 18001, ISO 22001?
ISO 9001 : Quality Standard Management
ISO 14001 : Environmental Standard Management
ISO 18001 : Safety & Health Standard Management
ISO 22001 : Hazop Standard Management
84.What is HACCP?
HACCP : Hazard Analysis Critical Control Point
85.What is OHSAS?
OHSAS : Occupational Health & Safety Assessment Series
86.Why one liter of water is equivalent to one kilogram of water at room temperature?
Because of at normal room temperature is between 25°C and 35°C at plant operating condition and the variation in weight Vs Liter of water is negligible compared to volume.
87.What is room temperature?
The temperature at prevailing working area
88.What is calibration?
The demonstration that a particular instrument or device produces results within specified limits by comparison with results produced by a reference or traceable standard over an appropriate range of measurements
89.What is the maximum time allowed after cleaning with water as last rinse?
Equipment should not be left with water it after cleaning. The last step of the cleaning procedures involve drying with solvent or flushing with nitrogen, thus ensuring that there is no opportunity for microbial growth.
90.What is the efficiency of the High Efficiency Particulate Air (HEPA) filter?
This type of air filter can remove at least 99.97% particles in air up to 0.3μm in diameter.
91. What is the micron size of HEPA filter?
The micron size of HEPA filter is 0.3μm
92.Do you have any idea about schematic diagram of HVAC system?
- Fresh Air
- Filtering of Air with Pre filter
- Cooling & Heating coil
- Filtering of Air with Fine filter
- Filtering of air with HEPA filter, If required
- Suction of air through return ducts from the process area using some pre filters as per requirements
- Air is exhausted to atmosphere after filtration wherever required
- Portion of air then passes through a dehumidifier wherever dehumidifier is required In the mixing chamber, return Air & Fresh air get mixed
- Process repeats from
93.If two different products are manufacturing in two modules of one production block, then will you accept the common air handling unit for both pharma area? Write “Yes” or “No” with reason?
No, because of cross-contamination (if re-circulation of return air)
Yes, if 100% of fresh air is circulated through the respective area.
94.Why blending validation is required?
What quality parameters of product are considered for validation and what parameters of equipment are to be considered during validation? Because of to provide sufficient documented evidence to assure that the blending operation of product is capable of repeatedly and reliably producing a homogeneous material to meet established specifications when operated under defined standard conditions. The following Quality parameters are to be considered, but not limited:
a) Loss on Drying / Water content
b) Bulk density / tapped density
c) Residual solvent
d) Particle size
The following parameters are to be considered for the equipment during validation, but not limited:
a) Blender capacity
b) RPM of the blender
c) Occupancy of the blender
d) Number of individual batches to be taken for each blend
e) Mixing time
95.What is the formula for calculation of “Air changes per hour” during HVAC validation?
Air changes per hour= Total CFM of the blower/Filter x 60 Total room volume
96.During the performance qualification in the vacuum tray dryer, how many temperature probs used?
Total 16 to 24 temperature probes are to be kept during the performance qualification of the vacuum tray dryer (or number of probes specified in the protocol)
97.What is the formula for the calculation of “MACO” while cleaning between one API to another API?
MACO =Minimum therapeutic dosage of previous product X Minimum batch size of next product
Safety factor X Maximum therapeutic dosage of the next product
98.What is the limit for “Individual unknown Impurity” in API as per ICH Q2A?
The limit of the “Any individual unknown Impurity” is not more than 0.1%
99.What are the class-I solvents as per ICH Q3C?
Benzene – 2 ppm Carbon tetrachloride – 4 ppm 1,2-Dichloroethane – 5 ppm 1,1-Dichloroethene – 8 ppm 1,1,1-Trichloroethane – 1500 ppm
100.What is the abbreviation of CAS Number?
CAS Number : Chemical Abstract Service Number
101.What is the specific gravity of Methylene chloride?
Specific gravity of Methylene chloride is 1.308 g/ml
102.If equipment is cleaned with water, then finally it should be rinse with suitable solvent as per guidelines, why?
Because of the last step of the cleaning procedures involve drying with solvent or flushing with nitrogen, thus ensuring that there is no opportunity for microbial growth.
103.What is mean by “4M”?
“4M” means Man, Machine, Method and Material
104.If supposed your pharma area is class 100000, then what is the maximum light and sound level as per guidelines?
The light & sound level limits are given below for class 100,000 / ISO 8:
Light Level : Not less than 300 Lux
Sound Level : Not more than 80 decibels
105.Write the classification of contaminants in clean rooms?
Substance:
Physical : Dust, Dirt, Grit, Fiber, Lint & Fly ash
Chemical : Organic compound, Inorganic salts, vapor, mist, fume & smoke Biologic : Bacteria, Fungus, Spore, Pollen, Virus, Human skin & cells
Energy:
Energy : Thermal, Light, Electromagnetic (EMI), Electrostatic (ESD), Radiation & Electrical
106.What is mean by “Clean-in-Place” and “Clean-out-Place”?
Clean-in-Place: The cleaning of large pieces of equipment may be performed in the equipments permanent location. Generally, in a configuration very similar to that in which it is utilized for production. This procedure widely known as Clean-in-Place (CIP)
Clean-out-Place: The smaller items are frequently transported to a designated cleaning or washing area where the cleaning procedures is performed. This practice is known as clean-out -place (COP)
107.What is the name of the instrument, which is used for measuring of vacuum (in Tars) during high vacuum distillation?
Macleod gauge
108.Is cGMP requirement only for personnel in the manufacturing?
No, this requirement is for each and every employee of the organization who must know the relevant cGMP requirements in his/her area.
109.Why cGMP should be followed?
This is a regulation that each one of us is trained in cGMP and practices cGMP – It minimizes the possibilities of any errors caused by subjectivity.
– It makes you do your job right the first time and every time.
110.For which areas do we have SOPs?
We have SOPs for the following areas:
– Quality Assurance
– Quality Control
– Production
– Personnel
– Warehouse
– Safety & Environment
– Engineering
– Estate Management
– Info tech
111.Why do we conduct trainings?
It brings awareness and helps us in becoming competent.
112.What is personal hygiene?
Each personal should:
– Wear clean uniform
– Take bath daily
– Report illness or injury
– Be medically fit
– Develop good hygiene habits
113.What are cGMP requirements for building and facilities?
Following are the cGMP requirements:
– Suitable size, construction and location
– Facilitate cleaning, maintenance and proper operation
– Adequate space
– Defined areas of adequate size
– Water supply: continuous and of good quality
– Power supply: continuous
– Adequate lighting, ventilations, air filtration, plumbing sewage, toilet facilities
114. What is mean by designated area?
By designated area we mean:
– Specific area for a specific operation: e.g. packing operation shall be carried out only in packing room and not elsewhere.
115.How do we know that the gauges are ok?
Gauges are periodically calibrated and they bear the calibration status tag.
116.What is in-process control?
Monitoring the manufacturing process at different stages is called in-process control. In-process control of the process provides an acceptable and achievable level of built in quality assurance for the product. This is possible through appropriate GMP during all manufacturing steps.
Or
Checks performed during production in order to monitor and, if necessary to adjust the process and / or to ensure that the intermediate or API conforms to its specifications.
117. What is critical process parameter?
A process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired quality. Or
A process condition or material or a test when it is essential to maintain a predetermined rage in order to reproducibly meet the specification is called critical parameter. Critical parameters have direct impact on the quality of a product.
118.What precautions are to be observed while working in the powder processing room?
Following precautions should be observed while working in the powder processing rooms: – Absolute discipline w.r.t complete uniform
– Bunny suit, clean shoe covers, hand loves, snoot mask etc. and SOPs compliance – Positive pressure
– House keeping
– Avoid foreign objects (pens, pencils, tools etc.)
– Identification / status card on materials
– Stage slips on equipments
– Temperature (less than 25°C)
– Avoid extraneous contamination from dust, insects, micro-organism, foreign particles etc. – Check the condition of sieves used in multi mill and sifter
– Cleaning and calibration of weighing balances
– Usage of fresh, clean drums and poly bags for final packing.
119.What precautions do we take during storage of API?
- All APIs are stored under controlled conditions of temperature and humidity in their designated area
- Records of temperature and humidity are maintained on daily basis.
- House keeping is done on daily basis and records are kept for the same.
- Insects, pests and rodent control procedures are follows.
120.What is mean by the word “Quality”?
Quality is basically customer’s satisfaction through sensitivity.
Or
A measure of a product’s or service’s ability to satisfy the customer’s stated or implied needs.
121. Inspection can be of three types, what are those?
Inspections are three types:
– Study /test based inspection
– Facility based inspection
– Process based inspection
122.Define stability study and its necessity?
Stability study is defined as “stability testing is to provide evidence how quality varies with time under influence as: temperature, humidity & light”
– Establish re-test period for drug substance
– Establish shelf life for drug product
– Recommended storage conditions
123.Write the different types of stability study conditions as per ICH guidelines?
- General storage conditions:
- Name Temperature (°C) Relative humidity (%) Long term 25±2 60±5
- Intermediate 30±2 65±5
- Accelerate 40±2 75±5
- Storage in a Refrigerators:
- Long term 5±3 NA
- Accelerate 25±2 60±5
- Storage in a Freezer:
- Long term -20±5 NA
124.What do you mean by “Reference standard” and “Working standard”?
Reference Standard: A substance that has been shown by an extensive set of analytical tests
to be authentic material that should be of high purity. This standard may be obtained from an officially recognized source or may be prepared by independent synthesis or by further purification of existing production material.
Working Standard: A substance of established quality and purity, as shown by comparison to a primary reference standard, used as a reference standard for routine laboratory analysis
125. What is abbreviation of CTD?
“CTD” means Common Technical Document. This is addressed in the ICH guidelines in the section of “M” and in the part of “M4”
126.What do you mean by market complaint?
Any communication, written or verbal, received regarding the quality, packing directly from any traders or product manufacturer and marketing staff or any other such complaints shall be considered as a Market Complaint.
127.What is maximum time period for the sending of the final response to concerned customer regarding the market complaint?
Within 30 days or as specified in the Market compliant SOP
128.Describe the categories of the market complaints?
Market complaints are categorized into three types and are as follows:
Critical: Complaints related to suspected contamination, adulteration and mislabeling. Major: Complaints related to the product not meeting its pre-determined critical specifications and damage to primary packaging.
Minor: Complaints related to the product not meeting non-critical quality attributes, or damage to secondary packaging or shortages etc.
129.What are the types of non-compliances in the internal audit?
Non-compliances shall be categorized as follows:
Critical: Those findings that warrant stoppage of any further operations in the facility until the corrective actions have been completed.
Major: Those findings that require immediate corrective action plan and compliance although operations can be continued.
Minor: Those findings that require corrective action plan as agreed between the Auditee department Head and Quality Assurance.
130.How many phases are divided the performance qualification of purified water system and write the duration of each phase?
Performance qualification of purified water system is divided into the three phases and described below:
Phase-I: (a) Develop & finalize operating, preventive maintenance, sanitization procedures. (b) Demonstrate production & delivery of water of required quality.
(c) To finalize SOP on sanitization, Alert & Action limits.
(d) Phase – I shall be conducted for 30 days.
Phase-II: (a) Demonstrate consistent operation within established ranges.
(b) Demonstrate consistent production & delivery of water of required quality. (c) Phase – II shall be conducted for 30 days.
Phase-III: (a) Demonstrate extended performance.
(b) Ensure that potential seasonal variations are evaluated & treated.
(c) Phase-III shall be conducted for 10 – 12 months
131.What is the abbreviation of “TDP” and its contents?
“TDP” means Technical Data Package and shall contain the following contents, but not limited: ∙ Brief manufacturing process
∙ Solvents used in the manufacture
∙ Impurity profile
∙ Working standard profile (If any)
∙ Characterization data (if any)
∙ Specifications and test procedures of the supplier
∙ TSE / BSE free Certificate
∙ Stability studies/ Hold time data
∙ Storage conditions
∙ Packing details
∙ MSDS
∙ Certificate Of Analysis (COA)
∙ DMF Number (if any)
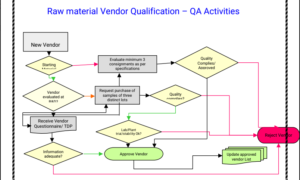
132.What are the QA activities during the vendor qualification for Key starting raw material, give flow chart?
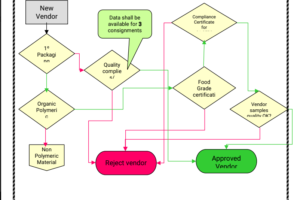
134.What are the QA activities during the vendor qualification for Primary packing material, give flow chart?
135.What do you mean by “worst case”?
A condition or set of conditions encompassing upper and lower processing limit and circumstances, within standard operating procedures, which poses the greatest chance of product or process failure when compared to ideal conditions. Such conditions do not necessarily induce product or process failure.
136.What do you mean by “performance qualification”?
The performance qualification documents describes the procedures for demonstrating that a system / piece of equipment can consistently perform & meet required specifications under routine operation and where appropriate, under worst case situations.
137.How will you close a market complaint?
(a) If satisfactory response obtained from complainant against our written reply (b) If the material is recalled
(c) If no response obtained from the complainant after 90 days (or specified in SOP) from date of our written reply.
138.Describe about swab and rinse sampling?
Swab: Areas which are reasonably accessible & hardest to clean can be evaluated, leading to level of contamination or residue per given surface area.
∙ Take the clean swab having surface area of 10mmX10mm
∙ Put the swab in the test tube containing 10 ml suitable solvent and squeeze the swab along the sides of the test tube to remove the excess of water from it.
∙ Identify the locations for swab sampling
∙ Take out the wet swab from the test tube without touching the tip of swab. ∙ Place the one side of swab over the identified location and apply it on the 10 X 10 sq. cm area first in vertical fashion without changing face of the swab
∙ Turn the swab to other side and apply it on the area in horizontal fashion covering all the areas
∙ Place the swab stick in to the test tube having 10 ml suitable solvent without touching the tip.
∙ Lave the test tube with the location
Rinse: Large area or parts of equipments which could not be swabbed should be rinse sampled or directly extracted by solvent. The areas which are not reasonably accessible for direct surface sampling have to be rinsed with suitable solvent.
∙ Use specified volume of suitable solvent for rinsing
∙ Rinse the identified locations using the following procedure
∙ Take specified quantity of suitable solvent in a graduated bucket
∙ Use a clean mug to splash the solvent in the reactor
∙ Close the bottom valve of the reactor
∙ Take solvent in the mug splash at all side of the reactor
∙ Attention to be applied particularly on the blind sides in the inside top of the reactor ∙ Splash the solvent at the agitator shaft and blades
∙ Open the bottom valve and collect the washed solvent in a clean bucket. Collect about 100 ml (specified quantity in the protocol) in a sample bottle from the bucket. Close the lid and label it properly.
∙ When more than one equipment is involved (equipment chain) for rinsing, suitable quantity of solvent shall be used and the rinse volume shall be measured.
139.What is the difference between specification and Limit?
Specification: A document giving a description of a starting material, packaging material, intermediate, bulk or finished product in terms of its chemical, physical & possibly biological characteristics. A specification normally includes description clauses & numerical clauses, the latter stating standards & permitted tolerances.
Or
It is the type of standard which is often referenced by a contract or procurement document. It provides the necessary details about the specific requirements.
Or
Lists of detailed requirements with which the products/ materials used or obtained during manufacture have to conform. They serve as a basis for quality evaluation.
Limit: The point, edge or line beyond which something cannot or may not be proceed. The boundary surrounding a specific area, bounds.
140.What are the possible causes for “Out of Specification”?
The following are the possible causes for out of specification:
∙ Test analysis error in QC Lab.
∙ Lab equipment malfunctioning or off-calibrated
∙ Production equipment malfunctioning or off-calibrated
∙ Operator/human errors
141.What is the clean room specifications for different classes (for >= 0.5 μm and >=5.0 μm particles) as per EU GMP/US 209E/ISO 14644-1/ Schedule M/ WHO GMP?
As per Schedule M:
Grade | Maximum permitted number of particles/m3equal or above | |||
At Rest | In Operation | |||
> 0.5 μm | >5.0 μm | > 0.5 μm | >5.0 μm | |
A | 3520 | 29 | 3500 | 29 |
B | 35200 | 293 | 352000 | 2930 |
C | 352000 | 2930 | 3520000 | 29300 |
D | 3520000 | 29300 | Not defined | Not defined |
As per USFDA (US 209E):
Clean Area | Maximum permitted number of particles/ft3 | Maximum permitted number of particles/m3 | ||
> 0.5 μm | >5.0 μm | > 0.5 μm | >5.0 μm | |
100 | 100 | 0 | 3520 | 29 |
1000 | 1000 | 7 | 35200 | 293 |
10000 | 10000 | 70 | 352000 | 2930 |
100000 | 100000 | 700 | 3520000 | 29300 |
As per ISO 14644-1:
Class | Maximum permitted number of particles/ft3 | Maximum permitted number of particles/m3 | ||
> 0.5 μm | >5.0 μm | > 0.5 μm | >5.0 μm | |
5 | 100 | 0 | 3520 | 29 |
6 | 1000 | 7 | 35200 | 293 |
7 | 10000 | 70 | 352000 | 2930 |
8 | 100000 | 700 | 3520000 | 29300 |
As per EU GMP:
Grade | Maximum permitted number of particles/m3equal or above | |||
At Rest | In Operation | |||
> 0.5 μm | >5.0 μm | > 0.5 μm | >5.0 μm | |
A | 3500 | 0 | 3500 | 0 |
B | 3500 | 0 | 350000 | 2000 |
C | 350000 | 2000 | 3500000 | 20000 |
D | 3500000 | 20000 | Not defined | Not defined |
WHO GMP 2002:
Grade | Maximum permitted number of particles/m3 | Maximum permitted number of particles/m3 | |
0.5 μm | 5.0 μm | Micro organisms | |
A | 3500 | 0 | <1 |
B | 3500 | 0 | 5 |
C | 350000 | 2000 | 100 |
D | 3500000 | 20000 | 500 |
142.What is the definition of document control?
Document control ensures that documents are reviewed for adequacy, approved for release by authorized personnel and distributed to and used at the location where the prescribed activity is performed.
143.What is the difference between controlled copy and un-controlled copy?
Controlled copy: A controlled copy is a formal copy of the latest, correct issue of a document; an identified issue of a document to an individual or location of record. The controlled copy is officially tracked, updated & destroyed to assure that it is current.
Uncontrolled copy: An informal copy of a document for which no attempt is made to update if after distribution; the document is marked “uncontrolled” and the user determines if the document is active prior to use.
144.What do you mean by re-validation?
A repeat of the process validation to provide an assurance that changes in the process/equipments introduced in accordance with change control procedures do not adversely affect process characteristics & product quality.
145.What are the system suitability parameters in the method validation? Tailing factor, Theoretical plates, Relative Standard deviation (RSD), Resolution, Retention time (RT), Relative retention time (RRT), Capacity factor
146.What is commissioning?
An engineering term that covers all aspects of bringing a system /sub-system to a position where it is regarded as being ready for use in pharmaceutical manufacture. Commissioning involves all the basis requirements of Installation qualification (IQ) & Operational Qualification (OQ)
147.When to qualify & validate?
Any aspect of, including significant change to the premises, the facilities, the equipment or the process, which may affect the quality of the product, directly or indirectly, should be qualified and validated.
148.What are the advantages of Swab sampling?
– Dissolves & physically removes sample
– Adaptable to a wide variety of surfaces
– Economical & widely available
– May allow sampling of a defined area
– Applicable to active, microbial & cleaning agent residues.
149.What are the advantages of Rinse sampling?
– Adaptable to on lime monitoring
– Easy to sample
– Non-intrusive
– Less technique dependent that swab
– Applicable for active, cleaning agents & excipients allow sampling of a large surfaces – Allows sampling of unique (e.g. porous) surface.
150.What are the different climate zones in the world?
World is divided into the Five climate zones and are given below:
Zone Name Conditions
I Temperate 21° C / 45% RH
II Subtropical & Mediterranean 25° C / 60% RH
III Hot & Dry 30° C / 35% RH
IVA Hot & Humid 30° C / 65% RH
VB Hot & Very Humid 30° C / 75% RH
151.What is a DMF?
A DMF is a package of proprietary information filed voluntarily by a company with the FDA. If it held by them in confidential closed files until such time as an FDA reviewer requests a review
of the DMF.
152.What are the types of DMF?
There are four types of DMF. They are referred to by their numbers in Roman numerals: Type-II: DMF is for companies who supply drug substances, drug products, intermediates & material used in their manufacture.
Type-III: DMF is for companies who supply packaging (container closure system) for human drugs & biologics.
Type-IV: DMF is for companies who supply excipients
Type-V: DMF is for companies who supply clinical services, sterile manufacturing etc. Type-I: Is an obsolete number once used for a category that no longer exists because it was found to easily fit into and overlap with the other four categories.
153.What are the possible reasons for the Non-conformities?
The following are the possible reasons, but not limited:
– Management attitude
– Ineffective documentation
– Lack of trained personnel
– Lack of co-ordination / co-operation within or among departments.
154.What do you mean by Critical Quality Attributes?
A critical quality attributes is a physical, chemical, biological or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.
155.What are the test parameters in the nitrogen gas validation?
Test parameters during validation and its frequency are given below:
a) Test for oil mists – Every 6 months once
b) Test for moisture content – Every 6 months once
c) Particulate count (Non-viable) – Every 6 months once
d) Sterility test (Aseptic area locations) – Every 6 months once
e) Bio burden test (Controlled area locations) – Every 6 months once * Perform the nitrogen gas sampling & testing for three consecutive days.
f) Purity of nitrogen gas (Based on manufacturer COA)
156.What do you mean by Non-conformity?
Non-conformity is non-fulfillment of a specified or implied requirement of the quality management system or of a quality work product.
157.Describe the method of testing for checking of MOC of SS material (Molybdenum test)?
Procedure:
i) Put one drop of electrolyte solution of molybdenum test kit on clean metal surface, which is to be tested.
ii) Switch on the detector and touch the metal tip of the detector on metal surface & carbon point in electrolyte solution.
iii) Do not pass the current for more than 3 to 4 seconds
iv) If the red color appears and is stable for more than 2 seconds then it can be concluded that MOC of the part being tested is SS-316.
v) If the solution remains colorless or green color appears then it can be concluded that MOC of the part being tested is SS-304.
vi) If the black color appears & is stable for more than 2 seconds then it can be concluded that MOC of the part being tested is SS-302.
158.What do you mean by production?
All operations involved in the preparation of an API from receipt of material through processing and packaging of an API.
159.Define Bio burden?
The level & type (i.e. objectionable or not) of micro organisms that can be present in raw materials, API starting materials, intermediates or APIs. Bio burden should not be considered contamination unless the levels have been exceeded or defined objectionable organisms have been detected.
160.What is the necessity of analytical method validation?
The principle purpose of analytical validation is to ensure that a selected analytical procedure will give reproducible and reliable results that are adequate for the intended purpose. It is thus necessary to define properly both the conditions in which the procedure is to be used & the purpose for which it is intended.
161.What is the solubility data as per any Pharmacopoeia?
Approximately volume of solvent in ml per gram of solute at
20° to 30° C
——————————————————————————
Very soluble : Less than 1
Freely soluble : From 1 to 10
Soluble : From 10 to 30
Sparingly soluble : From 30 to 100
Slightly soluble : From 100 to 1000
Very slightly soluble : From 1000 to 10000
Insoluble / practically soluble : More than 10000
Example: Very soluble means one gram of solute substance will require less than 1 ml of solvent.
162.What are the different types of safety factors used in the pharmaceutical industries? 1/10 to 1/100thof normal daily dose = Topical products
1/100 to 1/1000thof normal daily dose = Oral products
1/1000 to 1/10000thof normal daily dose = Inject able & Ophthalmic products 1/10000 to 1/100000thof normal daily dose = Research, investigational products.
163. What do you mean by reconciliation?
A comparison, making due allowance for normal variation, between the amount of product or material theoretically & actually produced/used.
164.What are the contents of Annual product quality review (APQR)?
Contents
1.0 Introduction
2.0 Number of Intermediates and APIs batches produced
3.0 Review of out put for all Isolated Intermediates and Finished Products
4.0Review of Critical Quality attributes of In-process, Isolated Intermediates and Finished Products
5.0Summary of changes made during the year with respect to equipment, Process, Specifications, and Methods, Raw materials and others.
6.0 List of Deviations and a brief description of deviations and action taken.
7.0List of customer complaints; Return goods and Recalled goods along with description and actions taken
8.0Number of Reprocessed and Reworks batches in all stages during the year 2009
9.0Review of Key starting materials and Primary packing materials and Rejections.
10.0 Review of Bio burden on product (for minimum of 3 batches) 11.0 Review on Stability studies and Summary
12.0 List of Out of specifications for Finished products
13.0 Review on Retained samples quality (Finished product)
14.0 Review on Validation packages (Process, Equipment, Procedure) 15.0 Status of Drug Master File (if any),Drug Master File new updates
16.0Details of special training provided to employees in case of Deviations or Complaints received regarding a particular product
17.0 Summary Report
165.Which solvents are categorized in the organic volatile impurities (OVI)?
Methylene chloride – 600 ppm
Chloroform – 60 ppm
1,1,2-Tri chloro ethylene – 80 ppm
1,4-Dioxane – 380 ppm
166.What do you mean by customer satisfaction?
Customer’s perception of the degree to which the customer’s requirements have been fulfilled Any customer wants three things:
– Good product
– Timely feedback
– Timely supply
167.What is the QMS?
It is the quality management system to direct and control an organization with regard to quality.
168. What do you mean by pre-determined acceptance criteria?
The criteria assigned, before undertaking testing to allow evaluation of test results to demonstrate compliance with a test phase of delivery requirement.
169.What is Master validation Plan (VMP)?
A document providing information on the company’s validation work program. It should define details of and timescales for the validation work to be performed. Responsibilities relating to the plan should be stated.
170.What do you mean by product recall?
Products recall means removal or withdraw of marketed material due to violation in laws & regulations as per regulatory authorities or not conforming to the customers’ specifications.
171. What do you mean by residual solvent?
These are the traces of the solvents left during the manufacturing of drug substances or drug products. Residual solvents are not completely removed by practical manufacturing techniques.
172.How can we avoid contaminations in warehouse?
Contamination can be avoided by:
– Proper segregation / identification of different RMs
– Following good house keeping procedures
– Having dedicated sampling tools for each RM
– Segregated rejected materials
– Segregated storage of penicillin RMs
– Separate sampling booth/enclosure for RMs
173.What is retention sample & why retention sample is preserved?
A part of the sample which is representative of the released batch of a finished product preserved beyond its shelf life.
It is preserved for future reference / reanalysis in cases of market complaints or development work or any other clarification about the released batch.
174.What will happen if cGMP are not followed?
Non-compliance to cGMP may lead to:
– Poor quality of product / services
– Batch failure
– Market complaints and product recalls
– Company’s reputation affected
– Business will be affected
– Regulatory action
– Injuries or accidents
– Equipment failures
175.What are the safety systems in the plant?
Some of the safety systems used in the plant are:
– Eye washer, safety showers
– Fire extinguishers
– Fire hydrants
– Face shields
– Goggles
– Helmets
– Nose masks
– Safety shoes
– Safety belts
– Hand gloves
– Training on safety rules and use of safety equipments.
176.Would you like to recommend any precautions while operating the equipment? Following precautions are recommended:
– Make sure that each equipment is numbered and its log is maintained. – Check whether the equipment is cleaned as per SOP
– Always follow product changeover procedure during product changeover – Take care of safety rules
– Monitor operating conditions
177.What are the contents of ICH Q7A GMP guidelines?
I.INTRODUCTION
A.Objective
B.Regulatory Applicability
C.Scope
II.QUALITY MANAGEMENT
A.Principle
B.Responsibilities of the Quality Unit(s)
C.Responsibility for Production Activities
D.Internal Audits (Self Inspection)
E.Product Quality Review
III. PERSONNEL
A. Personnel Qualifications
B. Personnel Hygiène
C. Consultants
IV. BUILDINGS AND FACILITIES
A. Design and Construction
B. Utilities
C. Water
D. Containment
E. Lighting
F. Sewage and Refuse
G. Sanitation and Maintenance
V. PROCESS EQUIPMENT
A. Design and Construction
B. Equipment Maintenance and Cleaning
C. Calibration
D. Computerized Systems
VI. DOCUMENTATION AND RECORDS
A. Documentation System and Specifications
B. Equipment Cleaning and Use Record
C. Records of Raw Materials, Intermediates, API Labeling and Packaging Materials
D. Master Production Instructions (Master Production and Control Records)
E. Batch Production Records (Batch Production and Control Records)
F. Laboratory Control Records
G. Batch Production Record Review
VII. MATERIALS MANAGEMENT
A. General Controls
B. Receipt and Quarantine
C. Sampling and Testing of Incoming Production Materials
D. Storage
E. Re-evaluation
VIII. PRODUCTION AND IN-PROCESS CONTROLS
A. Production Operations
B. Time Limits
C. In-process Sampling and Controls
D. Blending Batches of Intermediates or APIs
E. Contamination Control
IX. PACKAGING AND IDENTIFICATION LABELING OF APIs AND INTERMEDIATES
A. General
B. Packaging Materials
C. Label Issuance and Control
D. Packaging and Labeling Operations
X. STORAGE AND DISTRIBUTION
A. Warehousing Procedures
B. Distribution Procedures
XI. LABORATORY CONTROLS
A. General Controls
B. Testing of Intermediates and APIs
C. Validation of Analytical Procedures
D. Certificates of Analysis
E. Stability Monitoring of APIs
F. Expiry and Retest Dating
G. Reserve/Retention Samples
XII. VALIDATION
A. Validation Policy
B. Validation Documentation
C. Qualification
D. Approaches to Process Validation
E. Process Validation Program
F. Periodic Review of Validated Systems
G. Cleaning Validation
H. Validation of Analytical Methods
XIII. CHANGE CONTROL
XIV. REJECTION AND RE-USE OF MATERIALS
A. Rejection
B. Reprocessing
C. Reworking
D. Recovery of Materials and Solvents
E. Returns
XV. COMPLAINTS AND RECALLS
XVI. CONTRACT MANUFACTURERS (INCLUDING LABORATORIES)
XVII. AGENTS, BROKERS, TRADERS, DISTRIBUTORS, REPACKERS, AND RELABELLERS
A. Applicability
B. Traceability of Distributed APIs and Intermediates
C. Quality Management
D. Repackaging, Re-labeling, and Holding of APIs and Intermediates
E. Stability
F. Transfer of Information
G. Handling of Complaints and Recalls
H. Handling of Returns
XVIII. SPECIFIC GUIDANCE FOR APIs MANUFACTURED BY CELL CULTURE/ FERMENTATION
A. General
B. Cell Bank Maintenance and Record Keeping
C. Cell Culture/Fermentation
D. Harvesting, Isolation and Purification
E. Viral Removal/Inactivation steps
XIX. APIs FOR USE IN CLINICAL TRIALS
A. General
B. Quality
C. Equipment and Facilities
D. Control of Raw Materials
E. Production
F. Validation
G. Changes
H. Laboratory Control
I. Documentation
XX. GLOSSARY
178. What are the contents of the Drug Master File as per CTD Format?
Module-I:
1.1 Comprehensive Table of Contents
1.2 Application Form
1.3 Summary of Product Characteristics, Labeling and Package Leaflet
1.3.1 – Summary of Product Characteristics
1.3.2 – Labeling
1.3.3 – Package Leaflet
1.3.4 – Mock-ups and specimens
1.3.5 – SPCs already approved in the Member States
1.4 Information about the Experts
1.5 Specific requirements for different types of applications
1.5.1 Information for bibliographical applications under Art.4.8 (a) (ii) of Dir 65/65
1.5.2 Information for abridged applications under Art.4.8 (a) (iii) of Dir 65/65, 1st and 2n paragraph
Annexure: Environmental Risk Assessment
Module-2 (QOS):
2.1 CTD Table of Contents (Module 2 – 5)
2.2 CTD Introduction
2.3 Quality Overall Summary
2.4 Non-clinical Overview
2.5 Clinical Overview
2.6 Non-clinical Written and Tabulated Summary
– Pharmacology
– Pharmacokinetics
– Toxicology
2.7 Clinical Summary
– Bio pharmaceutics and Associated Analytical Methods – Clinical Pharmacology Studies
– Clinical Efficacy
– Clinical Safety
– Synopses of Individual Studies
Note: The contents of Quality Overall Summary are described below:
Quality Overall Summary
2.3. S Drug Substance
2.3. S.1 General Information
2.3. S.2 Manufacture
2.3. S.3 Characterization
2.3. S.4 Control of Drug substance
2.3. S.5 Reference standards or materials
2.3. S.6 Container closure system
2.3. S.7 Stability
2.3. R Regional Information
Module-3 (Quality):
Table of Contents
Body of Data
3.2. S Drug Substance
3.2 S.1 General Information
3.2.S.1.1 Nomenclature
3.2.S.1.2 Structure
3.2.S.1.3 General Properties
3.2. S.2 Manufacture
3.2.S.2.1 Manufacturers
3.2.S.2.2 Description of Manufacturing Process and Process Controls
3.2.S.2.3 Control of Materials
3.2.S.2.4 Controls of Critical Steps and Intermediates
3.2.S.2.5 Process Validation and/or Evaluation
3.2.S.2.6 Manufacturing Process Development
3.2. S.3 Characterization
3.2.S.3.1 Elucidation of Structure and other Characteristics
3.2.S.3.2 Impurities
3.2. S.4 Control of Drug substance
3.2.S.4.1 Specification
3.2.S.4.2 Analytical Procedures
3.2.S.4.3 Validation of Analytical Procedures
3.2.S.4.4 Batch Analyses
3.2.S.4.5 Justification of Specification
3.2. S.5 Reference standards or materials
3.2. S.6 Container Closure system
3.2. S.7 Stability
3.2.S.7.1 Stability Summary and Conclusions
3.2.S.7.2 Post approval Stability Protocol and Stability Commitment
3.2.S.7.3 Stability Data
179.What annexure are to be submitted while submitting the Drug Master File?
The following annexure are to be submitted during the initial submission wherever applicable: Annexure- 1 : Letter of Authorization
Annexure-2 : Letter of Agreement (Where the manufacturer is not intended holder of a certificate of suitability)
Annexure-3a : Letter of declaration of manufacture according to the presented dossier and to GMP rules for APIs for Human / Veterinary use.
Annexure-3b : Letter of declaration of manufacture according to the presented Dossier and to GMP rules and/or a quality assurance system.
Annexure-4 : Letter of declaration of willingness to be inspected according to the presented dossier and to the GMP rules.
Annexure-5 : Letter of declaration of manufacture regarding the use of material of human or animal origin including substances at risk of transmitting agents
of animal spongiform Encephalopathy.
Annexure-6 : Letter of commitment to provide samples of required by the EDQM.
180.Write the different parts of 21 CFR and details of different subparts of 21 CFR 211?
21 CFR Part 11 : Electronic records, Electronic signatures, Electronic copies of electronic record
21 CFR Part 58 : GLP for Non-clinical Laboratory studies regulation
21 CFR Part 210 : cGMP in manufacturing, processing, packing / holding of drug 21 CFR Part 211 : cGMP regulations for finished pharmaceuticals
21 CFR Part 820 : GMP regulations for Medical devices
Subpart A (General Provisions):
21 CFR Section 211.1 : Scope
21 CFR Section 211.3 : Définitions
Subpart B (Organization and Personnel):
21 CFR Section 211.22 : Responsibilities of Quality unit
21 CFR Section 211.25 : Personnel Qualification
21 CFR Section 211.28 : Personnel Responsibilities
21 CFR Section 211.34 : Consultants
Subpart C (Building and Facilities):
21 CFR Section 211.42 : Design & construction features
21 CFR Section 211.44 : Lighting
21 CFR Section 211.46 : Ventilation, Air filtration, Air heating & cooling
21 CFR Section 211.48 : Plumbing
21 CFR Section 211.50 : Sewage and refuse
21 CFR Section 211.52 : Washing and Toilet facilities
21 CFR Section 211.56 : Sanitation
21 CFR Section 211.58 : Maintenance
Subpart D (Equipments):
21 CFR Section 211.63 : Equipment design, size and location
21 CFR Section 211.65 : Equipment construction
21 CFR Section 211.67 : Equipment cleaning and maintenance
21 CFR Section 211.68 : Automatic, Mechanical and Electronic equipment 21 CFR Section 211.72 : Filters
Subpart E (Control of components and drug product container and closures):
21 CFR Section 211.80 : General Requirements
21 CFR Section 211.82 : Receipt and storage of untested components, drug product containers and closures.
21 CFR Section 211.84 : Testing and approval / rejection of components, drug product containers and closures
21 CFR Section 211.86 : Use of approved components, drug product containers and closures
21 CFR Section 211.87 : Retesting of approved components, drug product containers & closures
21 CFR Section 211.89 : Rejected components, drug product containers & closures 21 CFR Section 211.94 : Drug product containers and closures
Subpart F (Production and process controls):
21 CFR Section 211.100 : Written procedures and deviations
21 CFR Section 211.101 : Charge-in of components
21 CFR Section 211.103 : Calculation of yield
21 CFR Section 211.105 : Equipment identification
21 CFR Section 211.111 : Time limitation on production
21 CFR Section 211.115 : Re-processing
Subpart G (Packaging and labeling control):
21 CFR Section 211.122 : Materials examination and usage criteria
21 CFR Section 211.125 : Labeling issuance
21 CFR Section 211.130 : Packing and labeling operations
21 CFR Section 211.132 :Tamper-evident packing requirements for over-the-counter human drug products
21 CFR Section 211.134 : Drug product inspection
21 CFR Section 211.137 : Expiration dating
Subpart H (Holding and distribution):
21 CFR Section 211.142 : Warehousing procedures
21 CFR Section 211.150 : Distribution procedures
21 CFR Section 211.160 : General Requirements
Subpart I (Laboratory controls):
21 CFR Section 211.165 : Testing and release for distribution
21 CFR Section 211.166 : Stability Testing
21 CFR Section 211.167 : Special testing requirements
21 CFR Section 211.170 : Reserve samples
21 CFR Section 211.173 : Laboratory animals
21 CFR Section 211.176 : Penicillin contamination
Subpart J (Records and Reports):
21 CFR Section 211.180 : General requirements of records and reports 21 CFR Section 211.182 : Equipments cleaning and usage log
21 CFR Section 211.184 : Components, drug product containers, closures and labeling records
21 CFR Section 211.186 : Master production and control record
21 CFR Section 211.188 : Batch production and control record
21 CFR Section 211.192 : Production records review
21 CFR Section 211.194 : Laboratory records
21 CFR Section 211.196 : Distribution records
21 CFR Section 211.198 : Complaints files
Subpart K (Returned and salvaged drug products):
21 CFR Section 211.204 : Returned drug products
21 CFR Section 211.208 : Drug product salvaging
181.What parameters are to be considered during the analytical method validation of related substance or Residual solvents?
The following parameters are considered during the analytical method validation of related substances or Residual solvents:
PARAMETERS
1.Demonstration of precision
1.1 System precision / System suitability
1.2 Sample precision
2.Solution stability
3.Limit Of Detection (LOD)
4.Limit Of Quantification (LOQ)
4.1 Precision at LOQ level
4.2 Accuracy at LOQ level
4.3 Linearity at LOQ level
5.Accuracy
6.Linearity
7.Ruggedness
7.1 On the same column at spiked level
7.2 Variability from analyst to analyst
7.3 Variability from system to system
7.4 Variability from column to column
8.Robustness
8.1 Variability of column initial oven temperature
8.2 Variability of carrier gas flow rate
9.Different Batch analysis?
182.What parameters are to be considered during the analytical method validation of Assay?
PARAMETERS
1.System suitability
2.System precision
3.Sample precision
4.Solution stability and Mobile Phase stability
5.Assay Content
6.Accuracy
7.Linearity
8.Ruggedness
8.1 With same analyst, same system, same column
8.2 With same analyst, same system, but different column
8.3 With same analyst, same column, but different system
8.4 With same system, same column, but different analyst
9. Robustness
9.1 Effect of temperature variation on the column
9.2 Effect of flow variation
9.3 Effect of variation in mobile phase organic composition
9.4 Effect of pH variation in mobile phase
1.Different Batch analysis
183.What are the different conditions conducted during the forced degradation study?
The following conditions are conducted during forced degradation study:
a) Thermal degradation
b) Photo degradation (Direct sunlight)
c) Acid hydrolysis
d) Base hydrolysis
e) Oxidation
f) Ultra-violet exposure
184.What is clean zone and clean room?
Clean zone: A defined space in which the concentration of air borne particles is controlled to meet a specified air borne particulate cleanliness class
Clean room: A room in which the concentration of air borne particles is controlled and contains one or more clean zone.
185.What is As-built clean room (facility)?
A clean room (facility) that is complete and ready for operation, with all services connected and functional, but without equipment or operational personnel in the facility.
186. What is At-rest clean room (facility)?
A clean room (facility) that is complete and ready for operation, with all services functioning and with equipment installed and operable or operating, as specified, but without operating personnel in the facility.
187.What is Operational clean room (facility)?
A clean room (facility) in normal operation, with all services functioning and with equipment and personnel, if applicable, present and performing their normal work functions in the facility. Some other terms used in the clean rooms:
188.What is unidirectional and non-unidirectional air flow?
Unidirectional air flow: Airflow having generally parallel steam lines, operating in a single direction, and with uniform velocity over its cross section; previously referred to as “laminar” air flow
Non-unidirectional air flow: Air flow which does not meet the definition of unidirectional air flow, previously referred as “Turbulent or Non-Laminar” air flow.
189.What are the contents of US FDA GMP Guidelines?
I. Introduction
II. Organization and Personnel
A. Responsibilities of the Quality
Control Unit
B. Personnel Qualifications
C. Personnel Responsibilities
D. Consultants
III. Buildings and Facilities
A. Design and Construction Features
B. Lighting
C. Ventilation, Air Filtration, Air
Heating and Cooling
D. Steam, Gases, and Other Utilities
E. Plumbing, Washing, and Toilet
Facilities
F. Sewage and Refuse G.Sanitation
H. Maintenance
IV. Process Equipment
A. Equipment Design, Size, and
Location
B. Equipment Construction and
Installation
C. Equipment Cleaning and
Maintenance Procedures
D. Equipment Cleaning Methods
E. Validation of Equipment Cleaning
Methods
F. Clean in Place Methods
G. Automatic, Mechanical, Electronic,
and Computer
V. Control of Raw Materials
A. General Controls
B. Receipt, Sampling, Testing, and
Approval of Raw Materials
C. Use and Reevaluation of
Approved Raw Materials
D. Rejected Raw Materials
E. Control of Recovered Solvents,
Mother Liquors, and Second Crops
F. Process Water Quality
VI. Production and Process Controls
A. Written Procedures and
Deviations
B. Raw Material Weighing and
Measuring
C. Calculation of Yield
D. Equipment Identification
E. In-Process Controls,
Sampling/Testing of APIs and
Intermediates
F. Time Limits on Production of APIs
and Intermediates
G. Control of API Contamination
H. Blending of APIs and
Intermediates
VII. Packaging and Labeling Controls
A. Materials Examination and Usage
Criteria
B. API Label Issuance
C. Packaging and Labeling
Operations
D. API Packaging Materials
E. Expiration or Retest Dating
VIII. Holding and Distribution of APIs and Intermediates
A. Warehousing Procedures
B. Distribution Procedures
IX. Laboratory Controls
A. General Controls
B. Testing and Release for
Distribution
C. Stability Testing
D. Reserve Samples
E. Laboratory Animals
X. Records and Reports
A. General Controls
B. Equipment Cleaning and Use
Record
C. Raw Material, API Packaging
Materials, and Labeling Records
D. Master Production and Control
Records
E. Batch Production and Control Records
F. Production Record Review
G. Laboratory Records
H. Distribution Records
I. Complaint Files
J. Returned APIs and Intermediates
K. API and Intermediate Salvaging
XI. Validation
A. Process Validation Strategy
B. The Validation Protocol
C. Prospective Validation
D. Concurrent Validation
E. Retrospective Validation
XII. Change Control/Revalidation
A. Change-Control System
B. Change-Control Classification
XIII.Reprocessing/Reworking of APIs
and Intermediates
A. Reprocessing by Repeating a
Chemical Reaction
B. Reprocessing by Physical
Manipulations
C. Reworking of APIs and
Intermediates
XIV. Control of Chemical, Biological, and
Physical Contaminants
XV. APIs for Clinical Trials
A. Quality Assurance Measures
B. Quality Control Unit
C. Equipment and Facilities
D. Control of Raw Materials
E. Production and Process Controls
F. Process Validation
G. Change Documentation
H. Laboratory Controls
I. Documentation
190. What are the contents of South Africa (MCC) GMP Guidelines?
Chapter 1: Quality Management
Principles
Quality Assurance
Good Manufacturing Practice
Quality Control
Audits
Quality Evaluation Audits
Critical Procedures
Chapter 2: Organisation and Personnel
Principles
Responsibilities of Key Personnel
Legal Aspects
Qualifications
Training
Hygiene
Chapter 3: Premises and Equipment
Principles
Premises Equipment
Chapter 4: Materials Management
Principles
Purchasing
Receiving
Storage
Issuing
Chapter 5: Manufacturing
Principle
Validation
Dispensing
Manufacturing Operations
In-process Control
Contamination
Reprocessing
Chapter 6: Packaging
Principles
Component Issue
Packaging Operations
In-process Control
Contamination
Finished Product Release
Chapter 7: Quality Control
Principles
Responsibilities
Equipment
Personnel
Sampling
Testing
Standards, reagents
Documentation
Stability
Chapter 8: Documentation
Principles
Preparation, Issue and Use of Documents
Master Specifications
Master Manufacturing Instructions
Master Packaging Instructions
Batch Records (Starting Materials)
Batch Records (Packaging Materials) Batch Records (Manufacturing)
Batch Records (Packaging)
Other Procedures and Records
Analytical Records
Other Documentation Required
Chapter 9: Validation
Principles
Validation Master Plan
Validation Protocol
Validation Report
Qualification
Process Validation
Analytical Method Validation
Cleaning Validation
Computer System Validation
Validation of specific dosage forms
Chapter 10: Returned Goods
Principle
Procedures
Chapter 11: Complaints, Adverse Events, Recalls & Withdrawals
Principles
Complaints
Adverse Events
Recalls
Chapter 12: Contract Manufacture, Analysis and Servicing
Principles
Manufacture And/Or Packaging
Contract Analysis
Service Contracts
Chapter 13: Veterinary Medicines
Principles
General Requirements
Special Requirement
Chapter 14: Radio pharmaceuticals
Principles
Registration Requirements
Personnel
Premises and Equipment
Production and Handling of Radioactive Preparations
Packaging of Radio pharmaceuticals
Non-radioactive Kits
Distribution and Recalls
Quality Control
Chapter 15: Biological Medicines
Principles
Personnel
Premises and Equipment
Animal Quarters and Care
Documentation
The Possibility of Contamination
The Possibility of Infection
Where the Product Itself is an Infectious Agent
Starting Materials
Seed Lot and Cell Bank System
Operating Principles
Quality Control
Special Requirements for Final Testing
Waste Disposal
Chapter 16: Homeopathic Medicines
Principles
Premises
Documentation
Chapter 17: Medical Gases
Principle
General Requirements
Pipelines
Filling Areas
Preparation of Returned Cylinders
Filling
Lot Identification
Release
Storage
Chapter 18: Good Pharmaceutical Wholesaling Practice
Principles
General Requirements
Storage
Transport
Documentation and Control
Chapter 19: Electronic Data Processing
Principles
Responsibilities
Validation
Security
Chapter 20: Security Guidelines
Principle
Security Personnel
Entry to Site
Entry to Buildings
Internal Security
Chapter 21: Safety and Environmental Protection
Principles
Safety
Environmental Procedures
Chapter 22: Sterile Products
Introduction
Definitions
Facilities
Air Handling Systems
Sanitisation and Monitoring
Personnel Training
Manufacturing Requirements and Controls
Validation of Aseptic Process
Sterilisation Processes
Quality Control
Finishing of Sterile Products
Batch Release
Chapter 23: Isolator Technology
Principles
Definition of Terms
Isolator Design Principles
The Siting of Isolators
Factory Acceptance Test (FAT)
Installation Qualification (IQ)
Operational Qualification (OQ)
Process Qualification (PQ)
Microbiological Monitoring
Sanitisation of Materials
Gas Sterilisation of Isolator Systems
Chapter 24: Aerosols & Metered Dose Inhalers
Principles
General
Premises & Equipment
Production & Quality Control
191. What are the contents of Korean Drug Master File (KDMF)?
Section Contents
1.0 Facilities of Manufacturing site
1.1 Drawings of the whole site plan (each production, laboratory, storage area and
other accessory facilities for production should be labeled for area names, gates,
and corridor)
1.2 A simple plan showing the classification of the rooms (e.g. Class I, II, III, IV) using
different colors according to cleanliness
1.3 Schematic Drawings of air-ventilation system
1.4 Schematic Drawings of Compressed air system
1.5 Schematic Drawings of water system including sanitation
2.0 Data on physico-chemical properties and stability
2.1 Data on physico-chemical properties
2.1.1 Origin, finding and developmental history (including when, where, from
what and who extracted, isolated or synthesized the drug, what became the
fundamental source of such finding and when and where the non-clinical
studies and the clinical trial started)
2.1.2 Data on structure elucidation, physico-chemical properties and biological
properties
2.1.3 In case any patent has been acquired in Korea or foreign countries, data
including a copy of the patent register, etc.
2.2 Data on stability
2.2.1 As data in conformity with the “Guidance on Stability Test of Drugs, etc.” as
stipulated by the FDA Commissioner, specific analytical methods shall be
described and raw data shall be attached.
2.2.2 Test Methods
2.2.2.1 For drug substances, long-term stability data and stressed
condition stability data shall be submitted.
2.2.2.2 Stability data under the accelerated conditions may be submitted
in place of the long-term stability data.
3.0. Data on the manufacturing processes, packaging, containers, cautions in handling, etc.
3.1 Manufacturing processes
3.1.1 The overall manufacturing process flow shall be described in detail, and
such description shall include matters relating to in-process controls by
each manufacturing step. And data on starting materials, solvents,
reagents, etc. used in each manufacturing process, such as synthesis
(fermentation), isolation, purification, etc. shall be attached.
3.1.2 Detailed descriptions of key intermediates and information on their
specifications and test method shall be attached.
3.2 Container Closure System (Packaging & Containers)
3.2.1 A description of packaging methods to assure stability and discussion on
the suitability of the specific container closure system with respect to, for
example, choice of materials, shall be specifically described.
3.2.2 Market container system (packaging at release) shall be described
3.3 Cautions in Handling
3.3.1 Containers shall be categorized into well-closed container, tight container,
a hermetic container and the like in order to assure stability, and specific
storage conditions (i.e., to be stored in a dark place at room temperature or
in the refrigerator at 2-3°C) shall be described together with the containers.
The shelf-life shall be set based on the stability data or any other data that
can be officially acknowledged
4.0 Data evidencing that manufacturing practice of the drug substance is in conformity with
the Korea Good Manufacturing Practice (KGMP), Annex 4 of the Enforcement Rule or
anything equivalent thereto or higher.
4.1 For this purpose, the document prepared in accordance with Form # 77 attached
to the Enforcement Rule, or a copy of the certificate certifying that the practice is
in conformity with Annex 4 of the Enforcement Rule (In case the drug substances
manufactured in a foreign country, the bulk GMP certificate issued by the
government of the manufacturing country) shall be submitted
5.0 Data on batch analysis for drug substances, analytical procedures, the solvents used,etc.
5.1 Batch analysis shall be the results on the three or more consecutive lots in
accordance with the applicable specification and test method
5.2 The analytical procedures shall mean the specifications and test methods for drug
substances or intermediates and the method as specified in the Korean
Pharmacopoeia or in the foreign official compendia acknowledged by the KFDA
Commissioner shall be described. For drug substances using any other methods
than ones described in official compendia, an evidencing data shall be attached
thereto
5.3 In case any organic solvent is used during the manufacturing process, data on the
type of the organic solvent and the justification of the usage and the residual limits
of the organic solvent in the final product, the actual residual amount and the test
method shall be attached thereto
6.0 Sample drug substances as necessary for the quality test
6.1 The sample drug substances for the test of three times shall be provided unless
otherwise justified
192. What do you mean by site master file (SMF) and write its contents as per MHRA or PIC/S guidelines?
The Site Master File is prepared by the manufacturer and contains specific information about
the quality assurance, the production and/or quality control of pharmaceutical manufacturing
operations carried out at the named site and any closely integrated operations at adjacent and
nearby buildings.
The Site Master File provides information on the manufacturer’s operations and procedures
that can be useful in the efficient planning and undertaking of a GMP inspection.
C.1.General Information
C.1.1.Brief information on the firm (including name and address), relation to other sites and,
particularly, any information relevant to understand the manufacturing operations
C.1.2.Pharmaceutical manufacturing activities as licensed by the Competent Authorities
C.1.3.Any other manufacturing activities carried out on the site
C.1.4.Name and exact address of the site, including telephone, fax and 24 hrs telephone
numbers
C.1.5.Type of actual products manufactured on the site and information about specifically toxic
or hazardous substances handled, mentioning the way they are manufactured (in
dedicated facilities or on a campaign basis).
C.1.6.Short description of the site (size. location and immediate environment and other
manufacturing activities on the site).
C.1.7.Number of employees engaged in the quality assurance, production, quality control,
storage and distribution
C.1.8.Use of outside scientific, analytical or other technical assistance in relation to
manufacture and analysis
C.1.9.Short description of the quality management system of the firm responsible for
manufacture
C.2.Personnel
C.2.1.Organization chart showing the arrangements for quality assurance, including production
and quality control
C.2.2.Qualification, Experience and Responsibilities of key personnel
C.2.3.Outline of arrangements for basic and in-service training and how records are maintained
C.2.4.Health Requirements for personnel in the production
C.2.5.Personnel Hygiene requirements, including clothing
C.3.Premises and Equipment
C.3.1.Simple plan or descriptions of manufacturing areas with indication of scale architectural
or engineering drawings are not required).
C.3.2.Nature of Construction and finishes
C.3.3.Heat Ventilation & Air Conditioning System (HVAC)
C.3.4.Handling of Toxic, Hazardous and Sensitizing Chemicals
C.3.5.Brief description of water systems (schematic drawings of the systems are desirable)
including sanitation
C.3.6.Maintenance (description of planned preventive maintenance programmes and recording
system
C.3.7.Brief description of major production and control laboratories equipment
C.3.8.Maintenance (description of planned preventative maintenance programmes and
recording system).
C.3.9.Qualification and calibration, including recording system. Arrangements for computerized
systems validation
C.3.10.Availability of written specifications and procedures for cleaning manufacturing areas
and equipment
C.4.Documentation
C.4.1.Arrangements for the preparation, revision and distribution of necessary documentation
for manufacture
C.4.2.Any other documentation related to product quality which is not mentioned elsewhere
(e.g. microbiological controls on air and water)
C.5.Production / Manufacturing
C.5.1.Brief description of production operations using, wherever possible, flow sheets and
charts specifying important parameters (see at Appendix the list of products
manufactured)
C.5.2.Arrangements for the handling of starting materials. Packaging materials, bulk and
finished products, including sampling, quarantine, release and storage
C.5.3.Arrangements for Reprocessing and Rework
C.5.4.Arrangements for the Handling of Rejected Materials & Products
C.5.5.Brief Description of General Policy for Process Validation
C.6.Quality Control
C.6.1.Description of the Quality Control system and of the activities of the Quality Control
Department Procedures for the release of finished products
C.7.Contract Manufacturers and Analytical Laboratories
C.8.Distribution, Compliance and Product Recall
C.8.1.Arrangements and Recording System for Distribution
C.8.2.Arrangements for handling of Complaints and Recalls
C.9.Short description of the Self Inspection system
193. Describe about the Purified water system and validation?
The purified water system consists of following pre-treatment before final production of
purified water.
1. Pre-Treatment
2. Reverse Osmosis (RO) Unit
3. Electro De-ionization (EDI) System
4. Ultra filtration, storage.
Pre-Treatment:
Raw water is supplied from the APIIC (Andhra Pradesh State Industrial Infrastructure Corporation Ltd.)
(Manjeera water) and is stored in raw water tank, then the raw water is chlorinated thereafter. The
chlorinated water is then transferred to the Pressure sand filter to remove the suspended solids and
sodium Meta-bisulphite is dosed in the filtered water to neutralise any trace of residual chlorine present
in the filtered water and to ensure complete de-chlorination. This de-chlorinated is water passed
through the softener to reduce the hardness and then passed through cartridge filter to remove the
suspended solids, which may escape from up-steam units.
Reverse Osmosis (RO):
The filter water is fed to the Reverse Osmosis Plant, and the water passes through three RO units. The
RO is a process for removing dissolved mineral salts, organic molecules and certain other impurities
from water by forcing water under increased pressure to pass through a semi permeable membrane,
from that RO system, water is fed to the EDI system.
Electro De-ionization (EDI) System:
Electro deionization (EDI) is a common sense evolution of conventional ion exchange technology. In
EDI, just as in conventional ion exchange, cations and anions in the feed water are exchanged for
hydrogen and hydroxyl ions in the ion exchange resins, producing de-mineralized water. The key
operational difference is that with EDI, the ion exchange resin is regenerated continuously, while with
conventional ion exchange, chemical regeneration is performed intermittently.
Continuous regeneration in EDI is achieved electrochemically by means of ion conducting membranes
and an imposed electric current. The hydrogen and hydroxyl ions necessary for regeneration are
formed in-situ, without the addition of chemical reagents, by means of the familiar water dissociation
reaction, sometimes called water splitting:
H2O=H++ OH-
With EDI, feed water is fed through ion exchange resin in the diluting chambers bordered by anion- and
cation-conducting membranes. At the same time, electrodes at each end of the unit impose an electric
potential which drives the water splitting reaction and causes the ions in the ion exchange resins to
migrate to the selectively permeable membranes, where they are transported into the adjacent
concentrating chambers. Once in the concentrating chambers, the ions are carried away by the
concentrate flow. Note that just as in conventional ion exchange, EDI benefits from the excellent mass
transfer and de-ionizing characteristics of modern ion exchange resins.
ULTRA FILTRATION SYSTEM:
The EDI permeate water passed through the series of seven ultra filtration membranes and will take
care of bacteria, pyrogens, Total Organic Carbon (TOC) etc. and is collected in Purified water Storage
Tank. From this storage tank the purified water is passed through UV lamp. This water is again
circulated and collected through return loop.

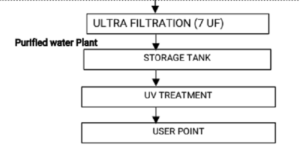
The validation of purified water generation shall be done in three phases. The purpose and sampling plan is
described as below:
The initial phase (phase-I) typically begins only after successfully completion of operational qualification.
The water generated during the second phases will be used for the manufacturing as long as the water
meets the specifications.
PHASE- I
During this phase daily samples were taken and analysed for chemical and microbiological quality.
Sampling should be after each step in the treatment process and from each point of use. The incoming
feed water will also be tested to verify the compliance with the specifications. The phase-I shall be
performed for a minimum period of 30days.
At the end of this phase alert and action limits will be established. These alert and action limits
will be used during phase-II and beyond.
The data obtained during phase-I should be used to develop the SOP and confirm that, the operational
SOPs are adequate.
Alert and Action limits shall be calculated based on the phase-1 study.
PHASE- II
This phase is to demonstrate that the system consistently operates within predetermined operating ranges
and delivers the water of the required quality (as specified) when operated in accordance with the SOPs.
The sampling plan will be the same as phase -I, and this activity will continue for 30 days after completion
of phase-I.
PHASE- III
This phase will be continued for 10months to verify the extended performance of system procedures on
the quantity and quality of water despite possible seasonal variations of feed water.
At the end of this phase the performance qualification is considered as completed and on going monitoring
will be established a continuous record of water quality. This activity is under progress. During this phase
purified water samples are collected daily from minimum one point of use, covering all points in a week.
PHASE | PRIMARY OBJECTIVES |
I | ∙ Develop & Finalize operating, preventive maintenance, sanitization procedures. ∙ Demonstrate production and delivery of water of the required quality ∙ To finalize the SOP on sanitization, Alert & Action Limit. |
II | ∙ Demonstrate consistent operation within established ranges. ∙ Demonstrate consistent production and delivery of water of the required quality. |
III | ∙ Demonstrate extended performance. ∙ Ensure that potential seasonal variations are evaluated and treated. |
Phase-I: samples shall be collected daily from all user points and tested for chemical and Microbiological
parameters. Chemical parameters like pH, TOC, Conductivity, Nitrates and Heavy metals were tested. For
Microbiology use high nutrient and low nutrient media to isolate the stressed organism.
The rationale of being low nutrient agar (R2A) being used during the validation of purified water was to
enumerate and isolate any adverse microorganisms, which are either slow growing or injured during
preliminary stages of water treatment. The bio-flim formed by the endogenous microorganism adsorbed
on to the surface is also indicative of surviving in low nutrient medium.
The samples tested using high nutrient agar were incubated at 30-35°C for 5 days and those tested using
low nutrient agar were incubated at 20-25°C for 7 days
Analyse the sample as per the Standard Operating Procedure. Phase-I will be started and duration of study
is minimum 30 days.
Phase-II: samples shall be collected daily from all user points and tested for Chemical and Microbiological
parameters. Chemical parameters like pH, TOC, Conductivity, Nitrates and Heavy metals were tested. For
Microbiology use high nutrient and low nutrient media to isolate the stressed organism.
The rationale of being low nutrient agar (R2A) being used during the validation of purified water was to
enumerate and isolate any adverse microorganisms, which are either slow growing or are injured during
preliminary stages of water treatment. The bioflim formed by the endogenous microorganism adsorbed on
to the surface are also indicative of surviving in low nutrient medium.
The samples tested using high nutrient agar were incubated at 30-35°C for 5 days and those tested using
low nutrient agar were incubated at 20-25°C for 7 days
For Microbiology use high nutrient and low nutrient media to isolate the stressed organism. Analyse the
sample as per the Standard Operating Procedure. Duration of Phase-II study is minimum 30 days.
Phase-III: samples are collected daily from minimum one point of use, covering all points in a week for Chemical and Microbiological parameters. For Microbiology use high nutrient media to isolate the stressed
organism. Analyse the sample as per the Standard Operating Procedure.
The samples tested using high nutrient agar was incubated at 30-35°C for 5 days. Duration of Phase-III
study is 10 months which also includes the seasonal variations.
194. What microbial & chemical parameters are to be considered during the nitrogen gas
validation?




